[AI Readability Summary]
Genome language models treat DNA and RNA sequences as learnable language. They support regulatory element recognition, variant effect prediction, RNA structure modeling, and generative sequence design while reducing heavy manual feature engineering and improving cross-task transfer.
Technical Specifications Snapshot
| Parameter | Details |
|---|---|
| Domain | Bioinformatics, genome foundation models |
| Primary Objects | DNA, RNA, regulatory sequences, variant sites |
| Modeling Paradigm | Transformer, sequence pretraining, multitask learning |
| Task Protocols | Classification, regression, sequence labeling, structure prediction, conditional generation |
| Data Sources | Genomic sequences, expression data, epigenomics, functional assays |
| GitHub Stars | Not provided in the source input |
| Core Dependencies | PyTorch, tokenizer/k-mer, attention, genomic benchmark datasets |
Genome language models have developed a clear task hierarchy
The original review can be summarized into eight task categories. The core logic is straightforward: first learn sequence semantics, then predict biological function, and finally move toward generative design. Compared with traditional bioinformatics pipelines, these models reduce manual motif engineering and rule construction while improving cross-task transferability.
 AI Visual Insight: This figure appears to be the review’s cover illustration. It emphasizes the panoramic role of genome language models in bioinformatics and typically connects three layers: tasks, model architectures, and benchmark datasets, rather than presenting specific experimental details.
AI Visual Insight: This figure appears to be the review’s cover illustration. It emphasizes the panoramic role of genome language models in bioinformatics and typically connects three layers: tasks, model architectures, and benchmark datasets, rather than presenting specific experimental details.
Regulatory element recognition is the most fundamental application scenario
This layer mainly answers the question, “What is this sequence segment?” It includes recognition tasks for promoters, enhancers, silencers, insulators, splice sites, and operon boundaries. Common formulations include binary classification, multiclass classification, and token-level labeling.
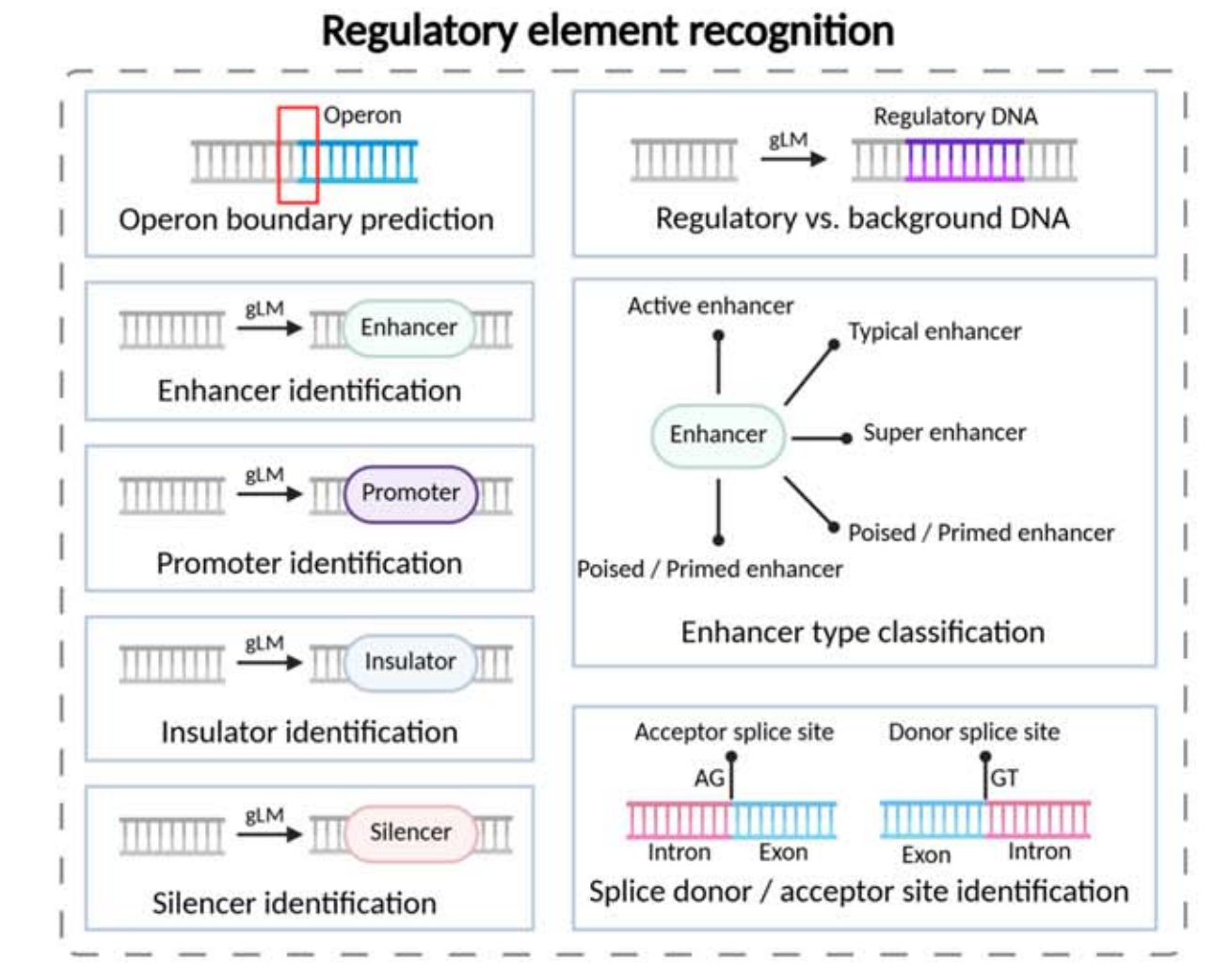 AI Visual Insight: The figure shows a functional decomposition of regulatory element recognition. In a typical setup, long genomic sequences are segmented and passed into a model, which outputs functional labels such as promoter, enhancer, or splice site. This illustrates the mapping from sequence patterns to biological regulatory semantics.
AI Visual Insight: The figure shows a functional decomposition of regulatory element recognition. In a typical setup, long genomic sequences are segmented and passed into a model, which outputs functional labels such as promoter, enhancer, or splice site. This illustrates the mapping from sequence patterns to biological regulatory semantics.
Quantitative regulatory activity prediction upgrades “whether it exists” to “how strong it is”
These tasks go beyond classification and predict continuous values such as promoter activity, enhancer activity, gene expression, and 3′ UTR regulatory strength. This setup is closer to experimental readouts and is better suited to testing whether a model truly understands regulatory grammar.
from typing import Dict
def predict_regulatory_task(seq: str) -> Dict[str, str]:
# Select the output head based on the task objective: classification or regression
if "TATA" in seq:
return {"task": "promoter_identification", "output": "classification"}
# Use a regression head when the target is expression strength
return {"task": "promoter_activity", "output": "regression"}This code snippet demonstrates the basic idea of attaching different task heads to the same sequence encoder.
Variant, interaction, and modification prediction are becoming high-value directions
Variant effect prediction is the most directly relevant direction for medical interpretation. The model compares reference and variant sequences and estimates the impact of an SNV or indel on transcription, splicing, chromatin accessibility, and pathogenicity. As a result, it is commonly used for pathogenicity scoring.
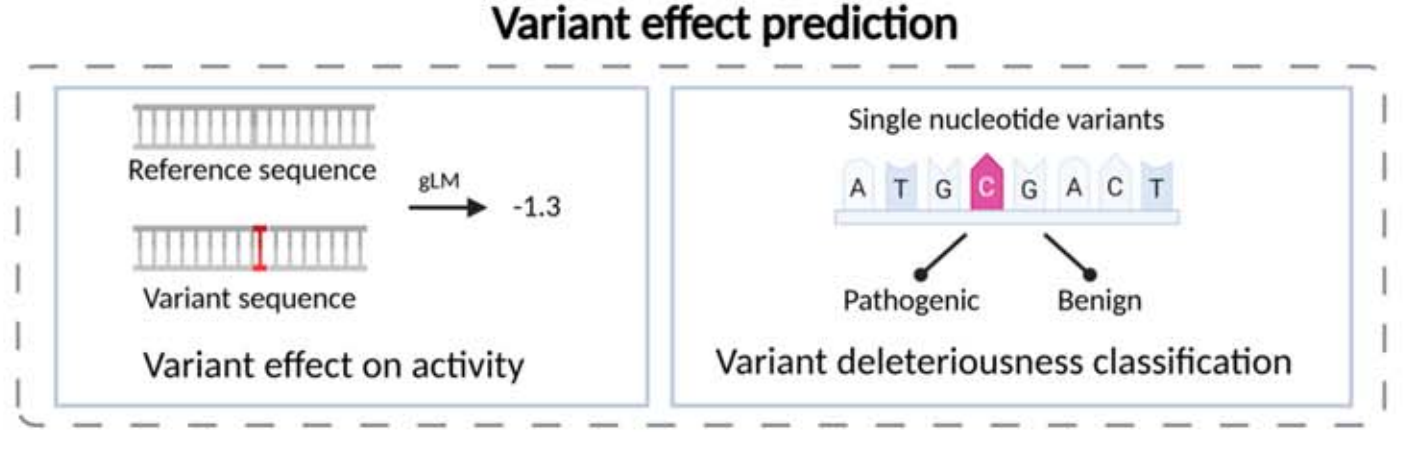 AI Visual Insight: This figure highlights the contrastive input mechanism between reference and mutated sequences. The model quantifies functional phenotypic changes through differential encoding or dual-tower comparison, which is an important technical route for clinical variant interpretation.
AI Visual Insight: This figure highlights the contrastive input mechanism between reference and mutated sequences. The model quantifies functional phenotypic changes through differential encoding or dual-tower comparison, which is an important technical route for clinical variant interpretation.
Chromatin and protein interaction modeling focuses on TF binding, histone marks, chromatin accessibility, and enhancer-promoter interaction. In essence, it uses sequence-only input to approximate epigenomics experimental outcomes.
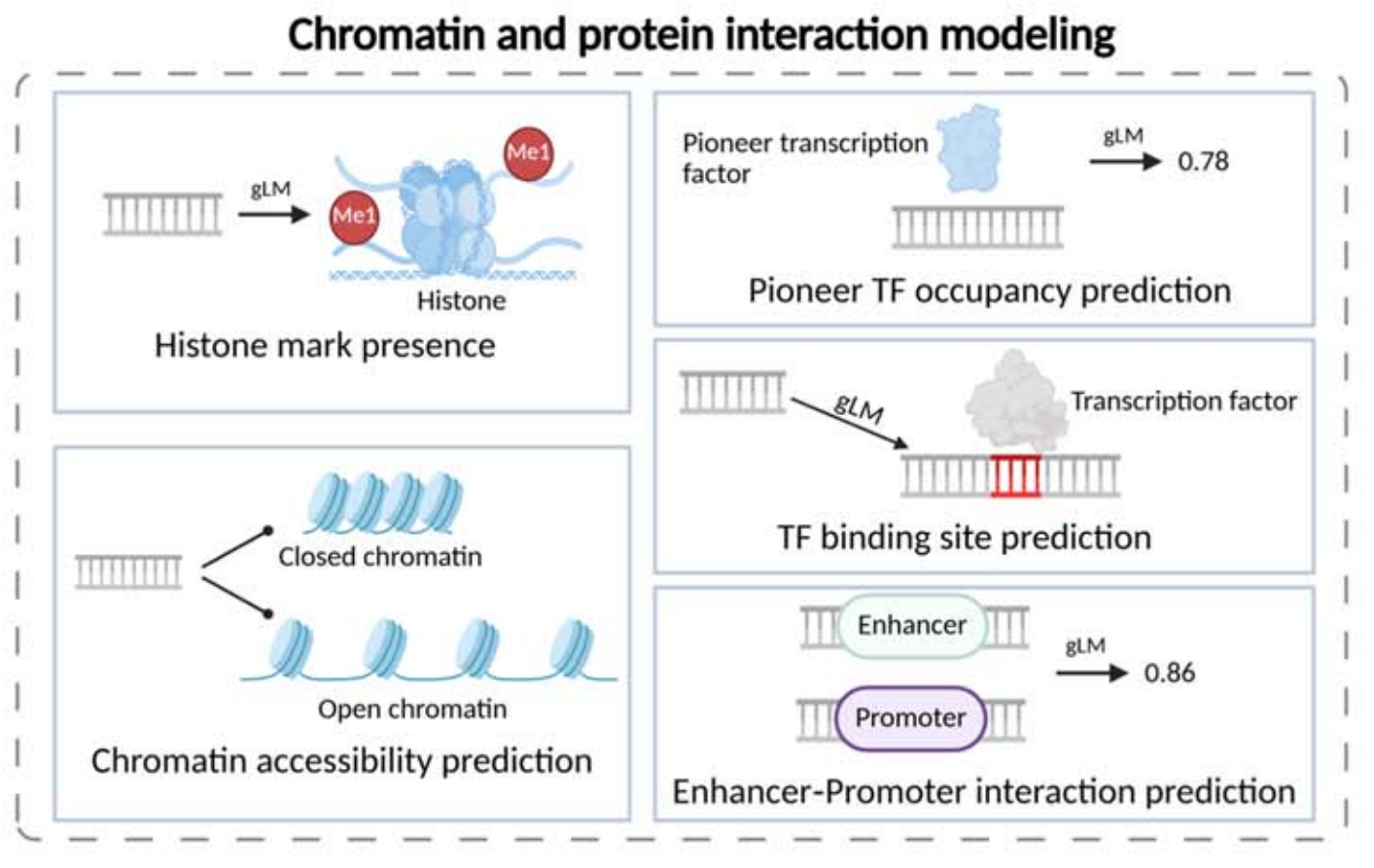 AI Visual Insight: The illustration typically maps DNA fragments to multi-label outputs, showing how a single model can jointly predict multiple transcription factor binding events, histone modifications, or open chromatin states. This reflects multitask modeling of epigenetic regulation.
AI Visual Insight: The illustration typically maps DNA fragments to multi-label outputs, showing how a single model can jointly predict multiple transcription factor binding events, histone modifications, or open chromatin states. This reflects multitask modeling of epigenetic regulation.
Molecular modification prediction and classification annotation expand the model’s generalization boundary
Modification prediction includes site detection for 5mC, 5hmC, 6mA, 4mC, and RNA m6A. Classification and annotation tasks cover species or lineage identification, coding potential, gene biotype, and lncRNA versus mRNA classification. This shows that foundation models can support not only regulation tasks, but also annotation and identification.
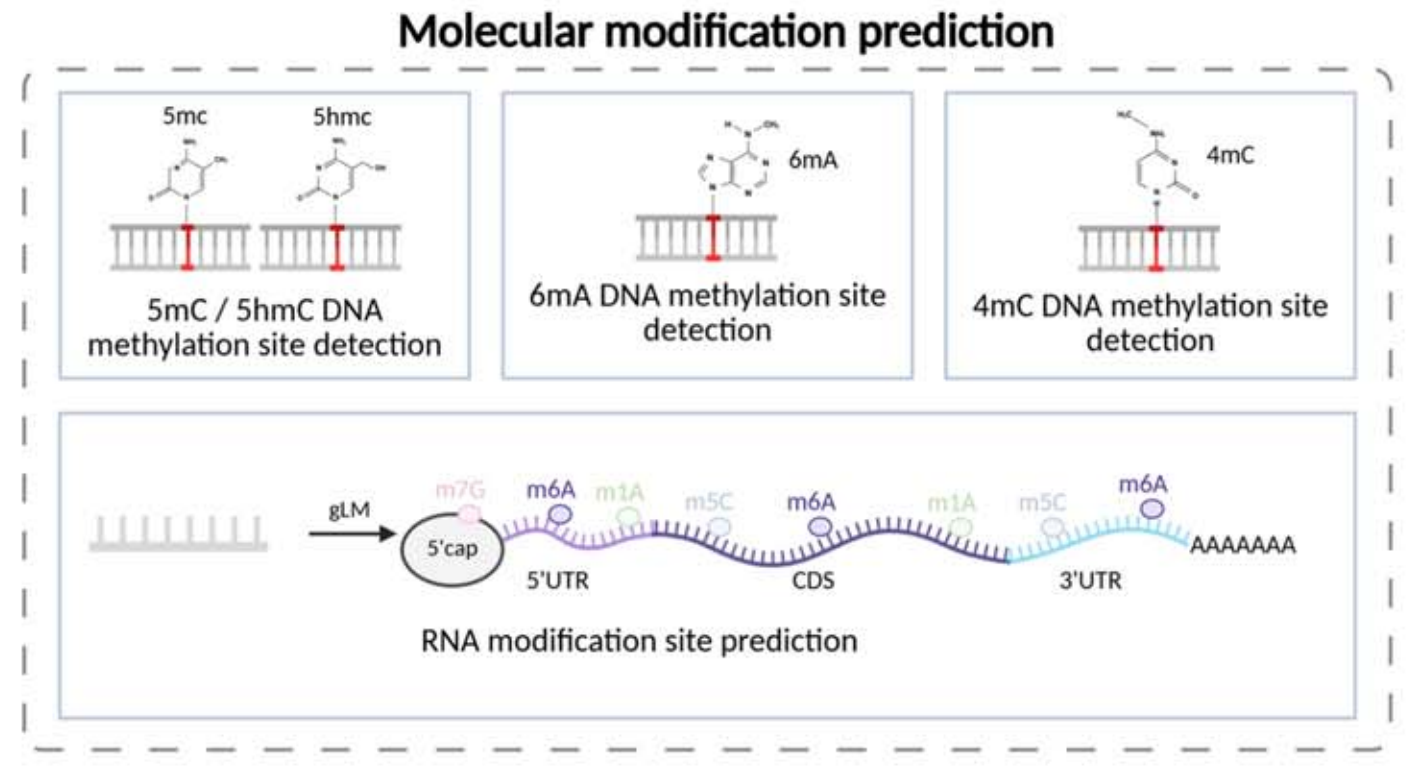 AI Visual Insight: This figure focuses on methylation and RNA modification site recognition. The typical input is a local nucleic acid window, and the output is a single-base modification label, highlighting the model’s sensitivity to microscopic chemical modification signals.
AI Visual Insight: This figure focuses on methylation and RNA modification site recognition. The typical input is a local nucleic acid window, and the output is a single-base modification label, highlighting the model’s sensitivity to microscopic chemical modification signals.
Generative design and RNA tasks represent the frontier of model capability
Generative sequence design shifts the goal from “understanding sequences” to “constructing sequences.” Targets include conditional generation of artificial genomes, promoters, and enhancers. Constraints may include species, GC content, sequence length, or expression strength.
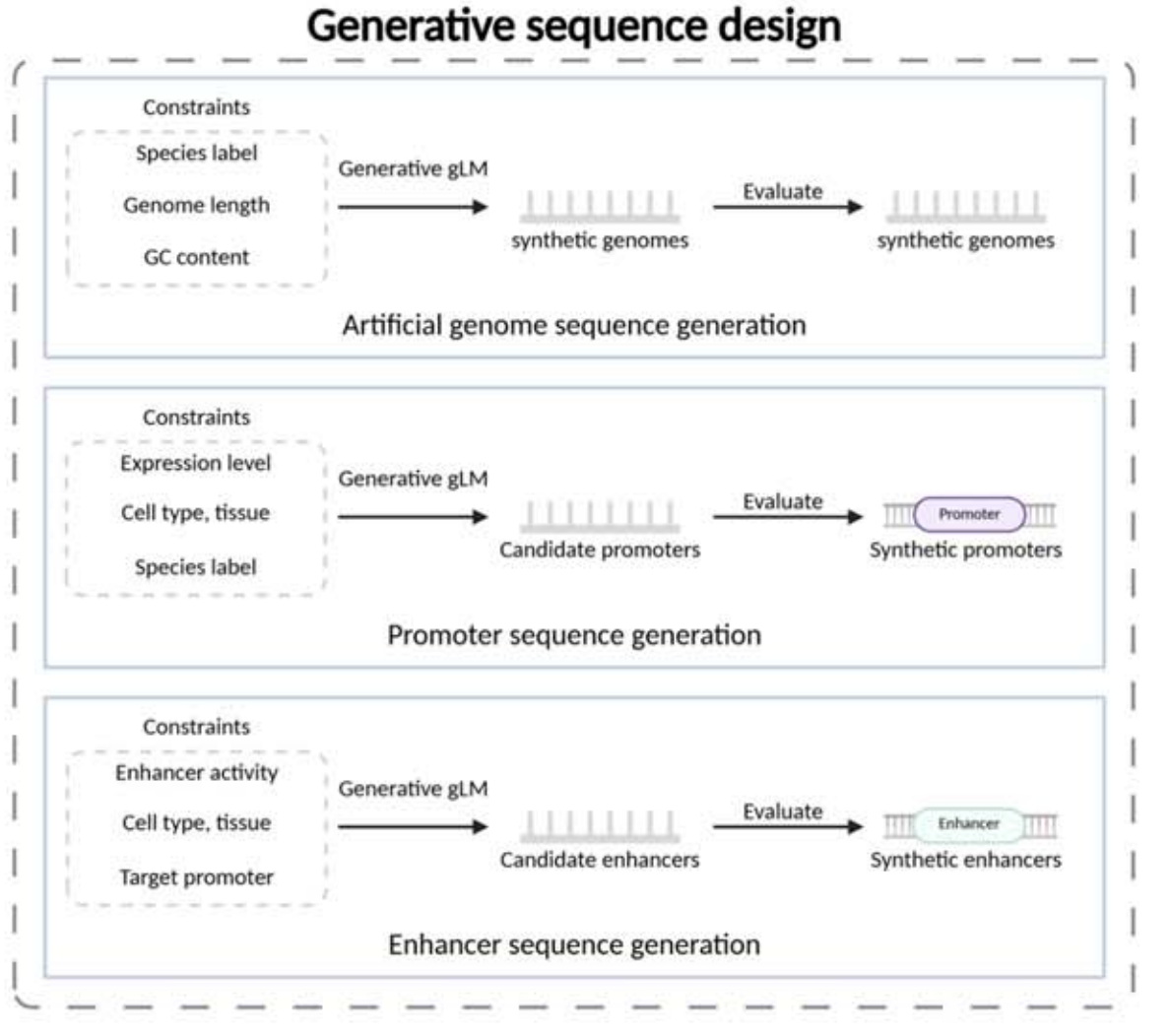 AI Visual Insight: This figure emphasizes a conditional generation framework in which the model receives functional constraints or species labels and produces new sequences that satisfy the target properties. It is especially relevant to promoter and enhancer design in synthetic biology.
AI Visual Insight: This figure emphasizes a conditional generation framework in which the model receives functional constraints or species labels and produces new sequences that satisfy the target properties. It is especially relevant to promoter and enhancer design in synthetic biology.
RNA tasks are more complex. Beyond expression prediction, they also involve secondary structure, tertiary contact maps, half-life, translation efficiency, RBP binding, and IRES recognition. Outputs are often matrices, dot-bracket notation, or continuous expression values rather than a single classification label.
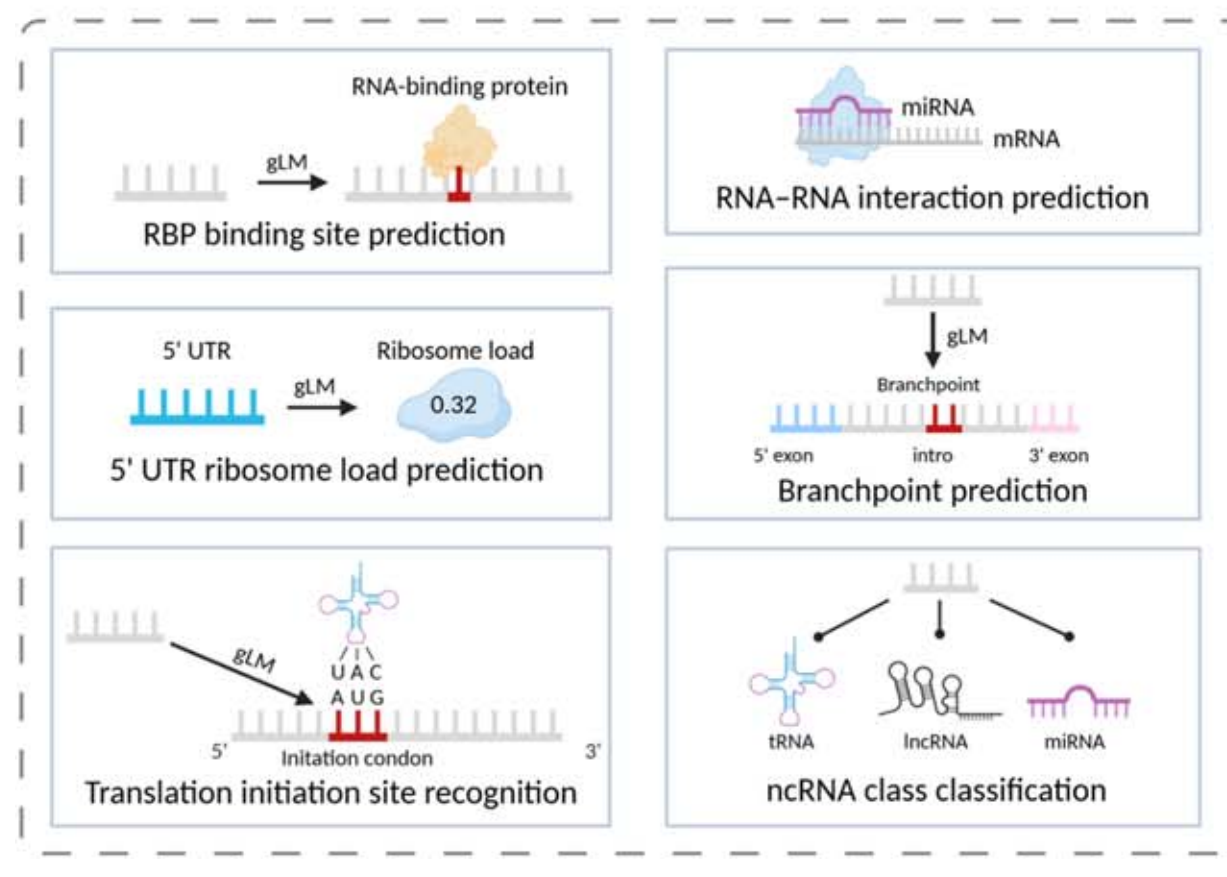 AI Visual Insight: This figure typically represents the diversity of RNA functional tasks, such as RBP binding, translation efficiency, and splice site recognition. It shows that RNA sequence modeling must handle both local motifs and long-range dependencies.
AI Visual Insight: This figure typically represents the diversity of RNA functional tasks, such as RBP binding, translation efficiency, and splice site recognition. It shows that RNA sequence modeling must handle both local motifs and long-range dependencies.
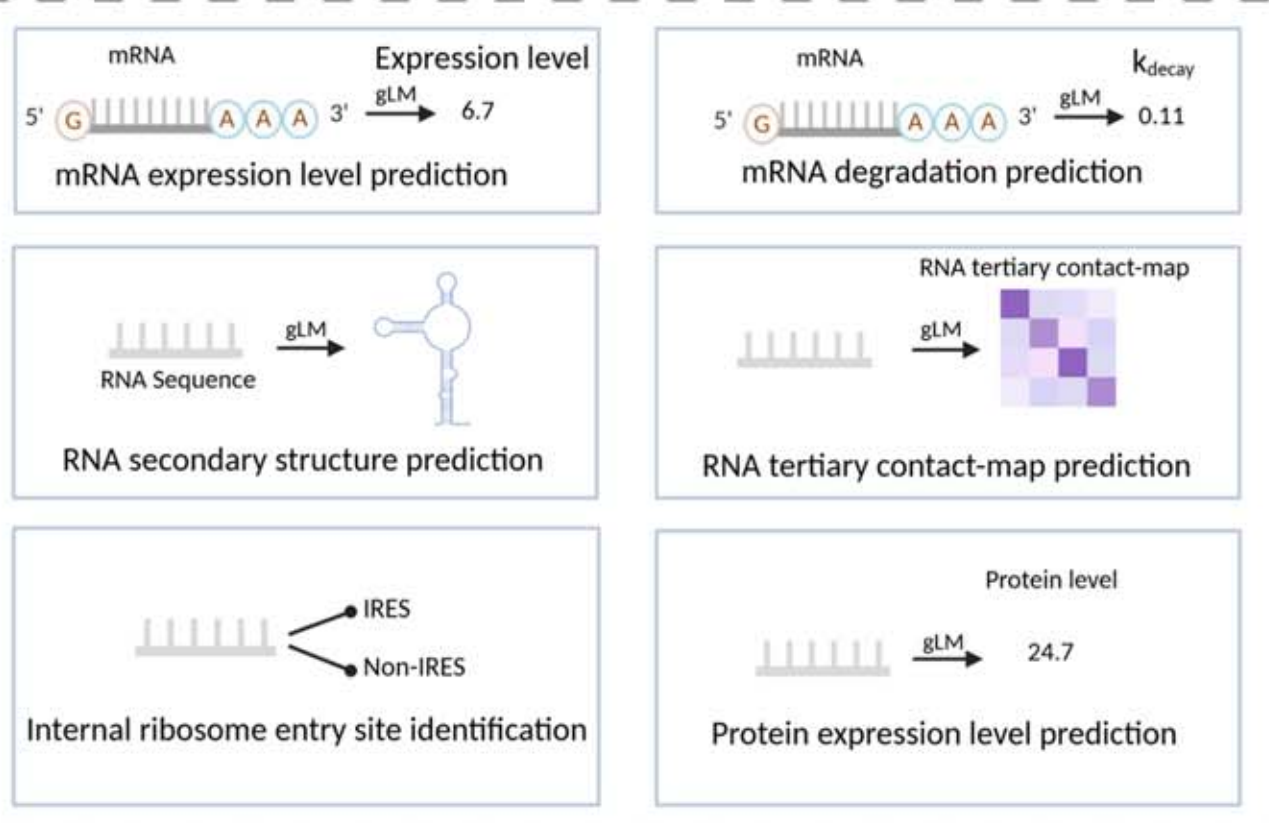 AI Visual Insight: This figure is more focused on RNA structure tasks and may include secondary structure pairing relationships or base-level contacts. It highlights the core challenge of inferring structural topology from linear sequence.
AI Visual Insight: This figure is more focused on RNA structure tasks and may include secondary structure pairing relationships or base-level contacts. It highlights the core challenge of inferring structural topology from linear sequence.
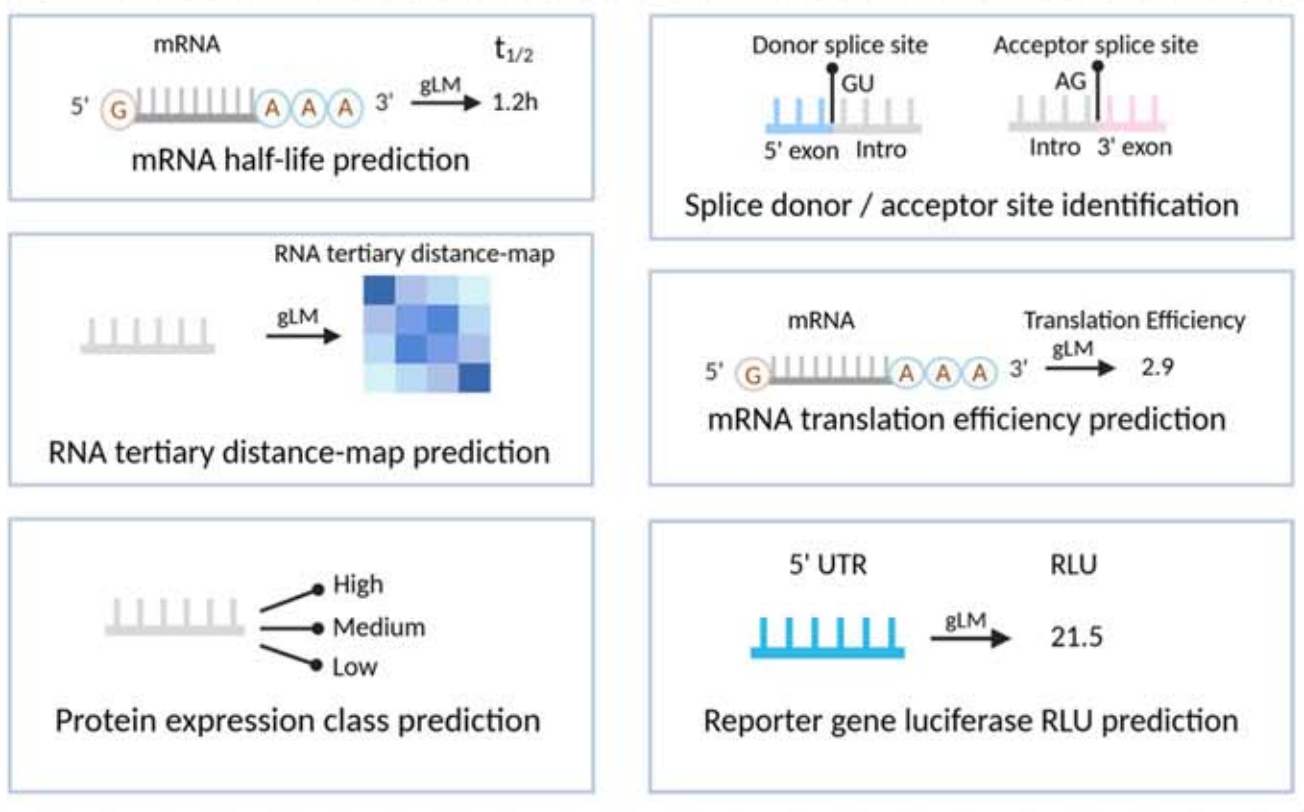 AI Visual Insight: This figure extends to tertiary contact map or distance map prediction, which means the model output is no longer a single value but a nucleotide-by-nucleotide relational matrix. This places extremely high demands on long-range modeling.
AI Visual Insight: This figure extends to tertiary contact map or distance map prediction, which means the model output is no longer a single value but a nucleotide-by-nucleotide relational matrix. This places extremely high demands on long-range modeling.
def compare_variant(ref_score: float, alt_score: float) -> float:
# Variant effect is often represented as the difference in functional score
delta = alt_score - ref_score
# A positive value indicates increased activity, and a negative value indicates decreased activity
return deltaThis snippet captures the most common scoring method used in variant effect prediction.
Benchmark design determines whether a model has real biological value
The original material included a benchmark section but did not expand the specific dataset list. Methodologically, a genomic benchmark should cover at least four things: task diversity, cross-species generalization, long-sequence modeling capability, and experimental label reliability.
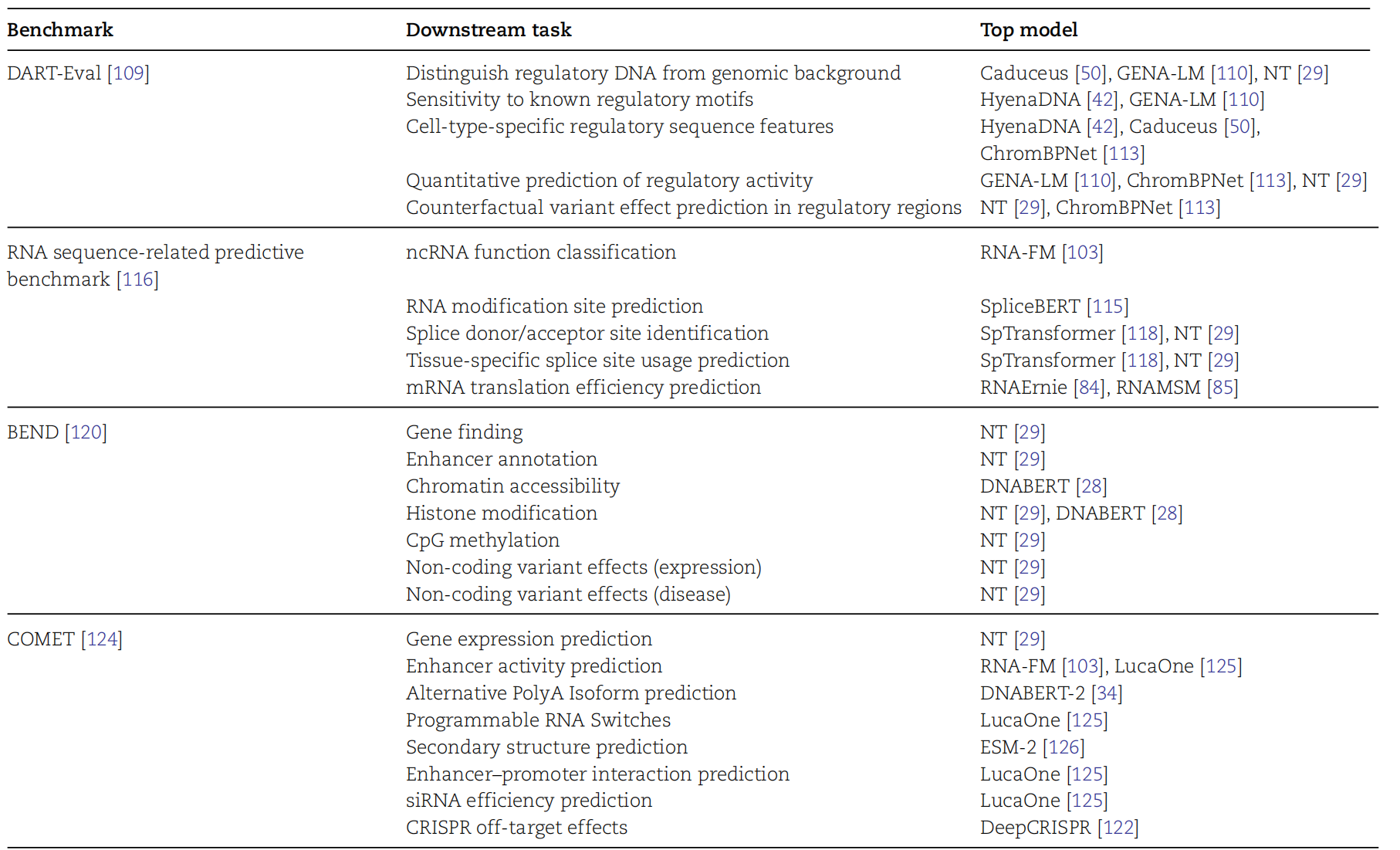 AI Visual Insight: This figure is likely a benchmark overview organized as a matrix across tasks, data modalities, and evaluation dimensions. It is used to compare model coverage in classification, regression, and structure prediction.
AI Visual Insight: This figure is likely a benchmark overview organized as a matrix across tasks, data modalities, and evaluation dimensions. It is used to compare model coverage in classification, regression, and structure prediction.
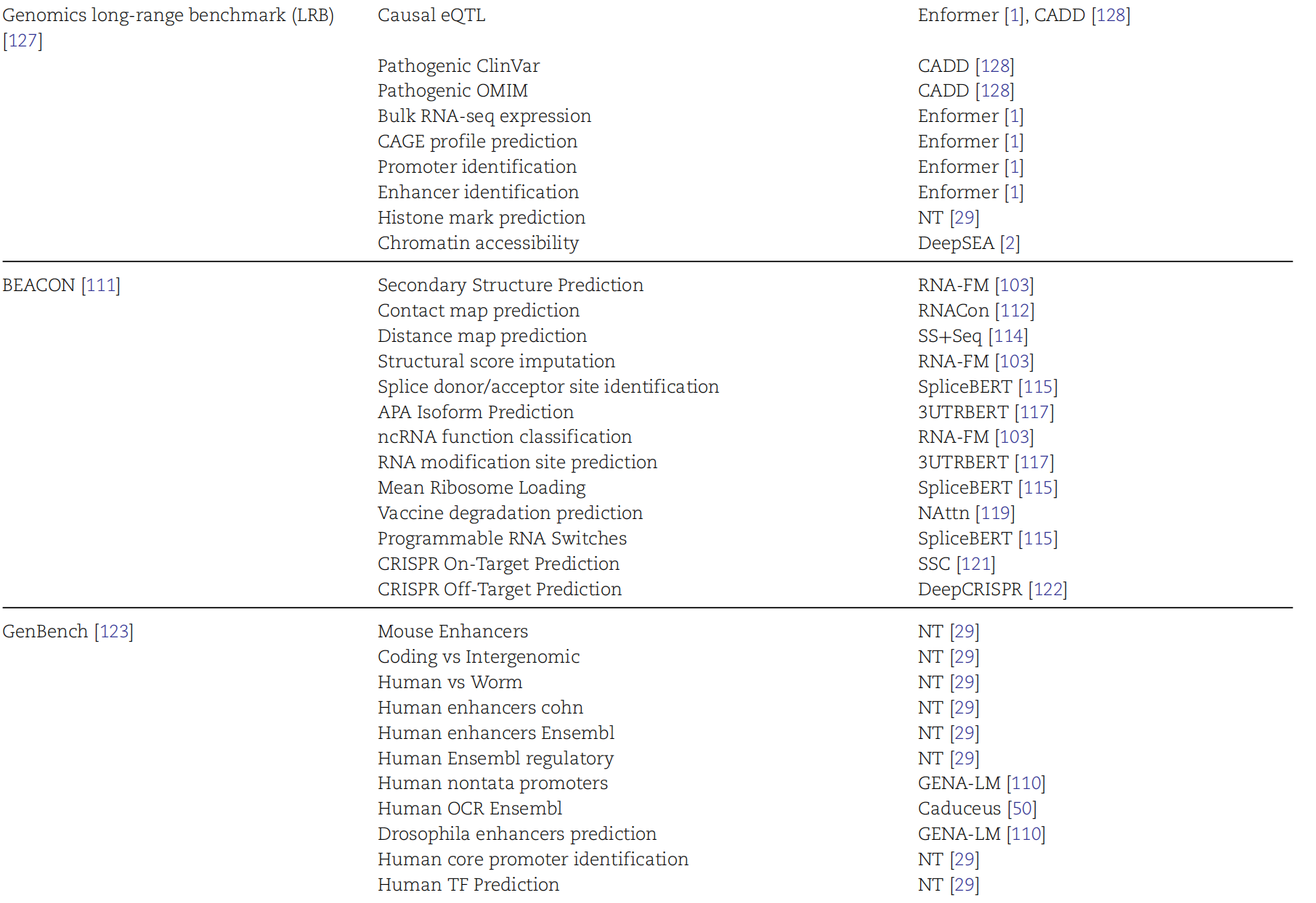 AI Visual Insight: This figure most likely shows the correspondence between benchmark datasets or task families, helping researchers determine whether a model is oriented toward DNA regulation, variant interpretation, or RNA structure.
AI Visual Insight: This figure most likely shows the correspondence between benchmark datasets or task families, helping researchers determine whether a model is oriented toward DNA regulation, variant interpretation, or RNA structure.
 AI Visual Insight: This figure looks more like an evaluation framework or model comparison view. It emphasizes unified input-output protocols and multidimensional metrics, both of which are essential for building reproducible experimental baselines.
AI Visual Insight: This figure looks more like an evaluation framework or model comparison view. It emphasizes unified input-output protocols and multidimensional metrics, both of which are essential for building reproducible experimental baselines.
The main thread in representative models is “longer context, stronger pretraining, and more specialized task heads”
Although the original text did not provide a named list of models, the evolution of this field generally follows three routes: k-mer or BPE sequence encoding, long-context Transformers, and multitask fine-tuning aligned with biological experimental readouts. Evaluation should not focus only on average scores. Interpretability and cross-dataset stability also matter.
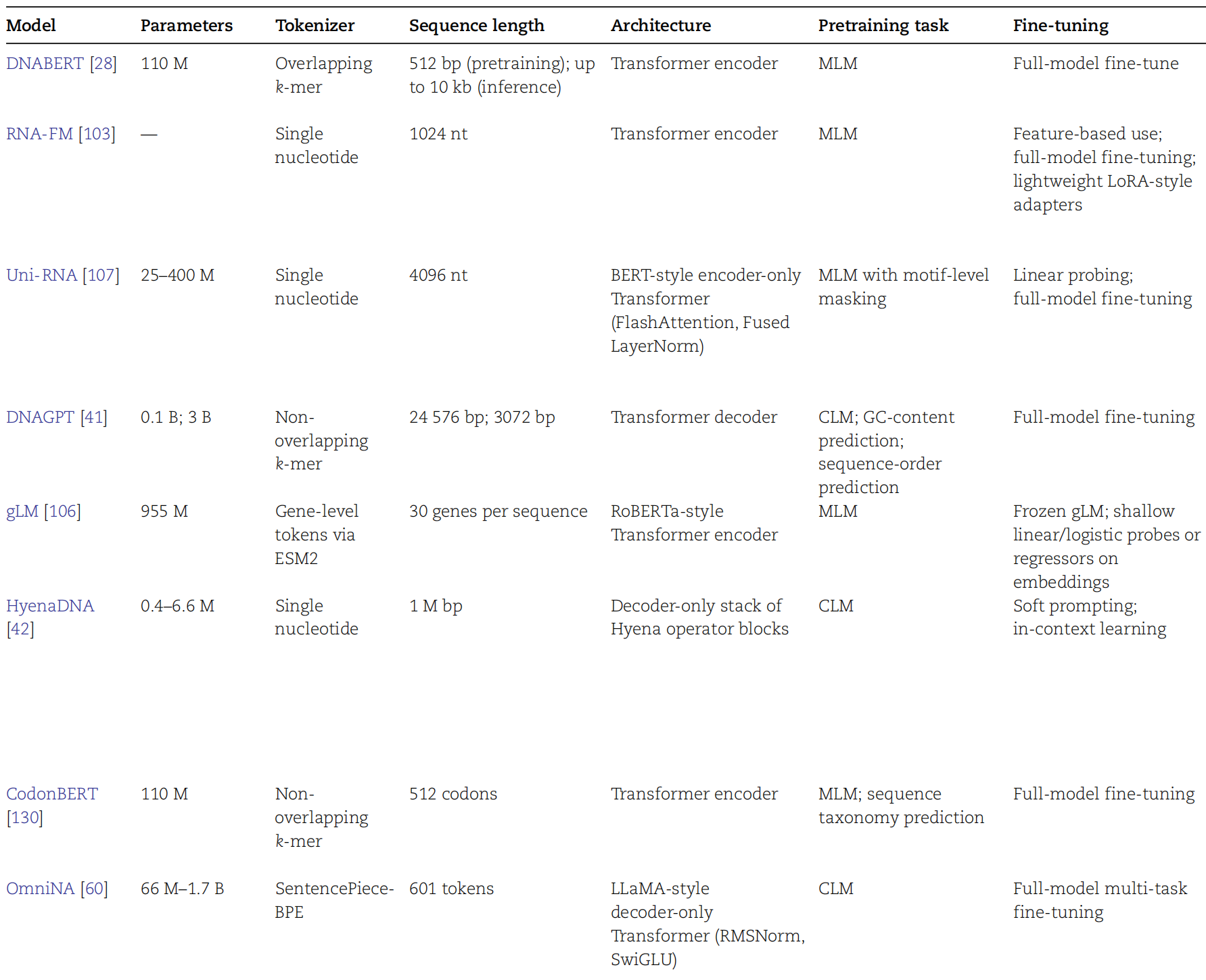 AI Visual Insight: This figure likely summarizes the landscape of representative model families, possibly grouped by year, architecture, or task domain. It shows the transition from specialized classifiers to general pretrained genome foundation models.
AI Visual Insight: This figure likely summarizes the landscape of representative model families, possibly grouped by year, architecture, or task domain. It shows the transition from specialized classifiers to general pretrained genome foundation models.
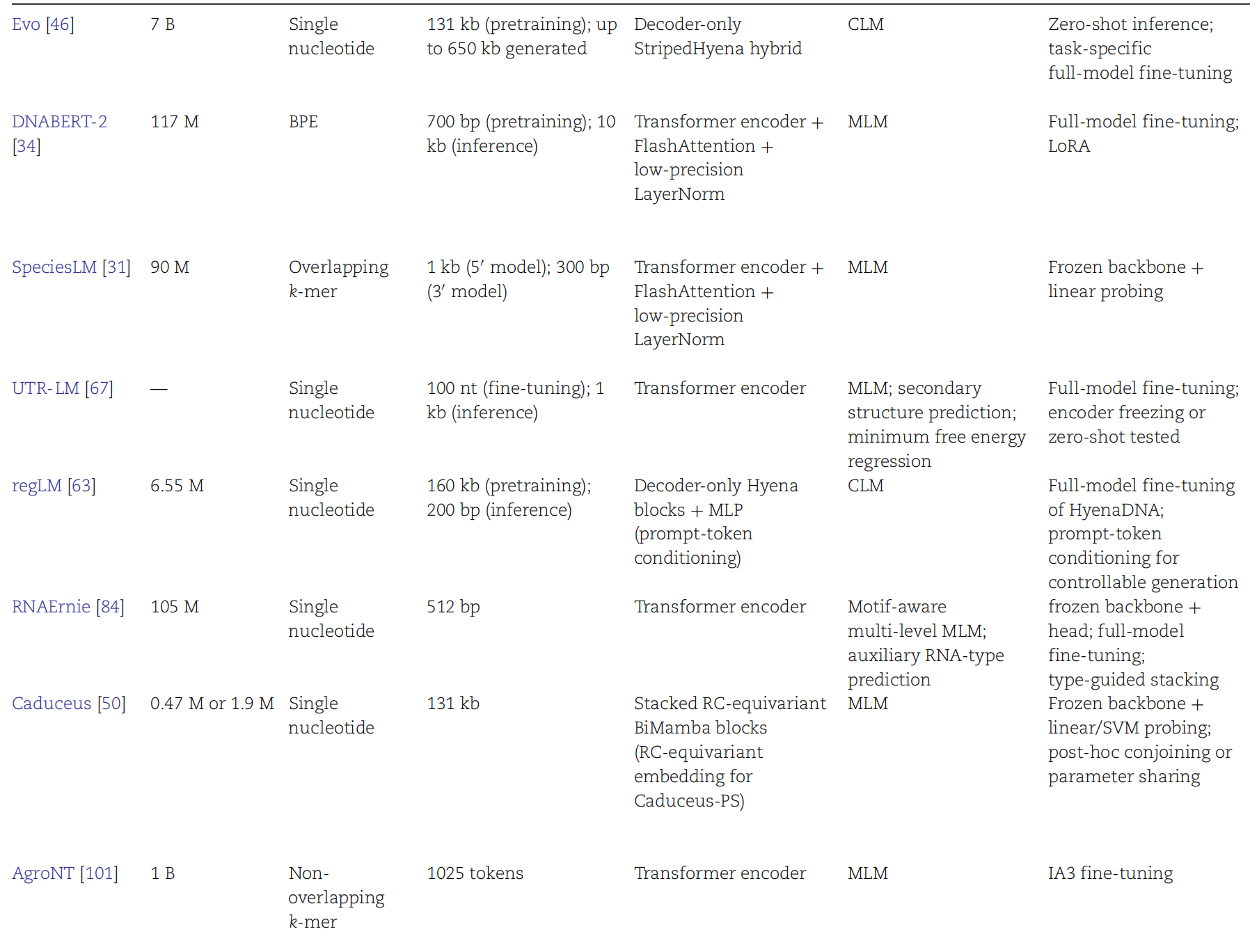 AI Visual Insight: This figure may compare input length, parameter scale, pretraining corpora, and downstream task coverage across models, making it easier to judge whether a model is better suited to long sequences or high-precision functional prediction.
AI Visual Insight: This figure may compare input length, parameter scale, pretraining corpora, and downstream task coverage across models, making it easier to judge whether a model is better suited to long sequences or high-precision functional prediction.
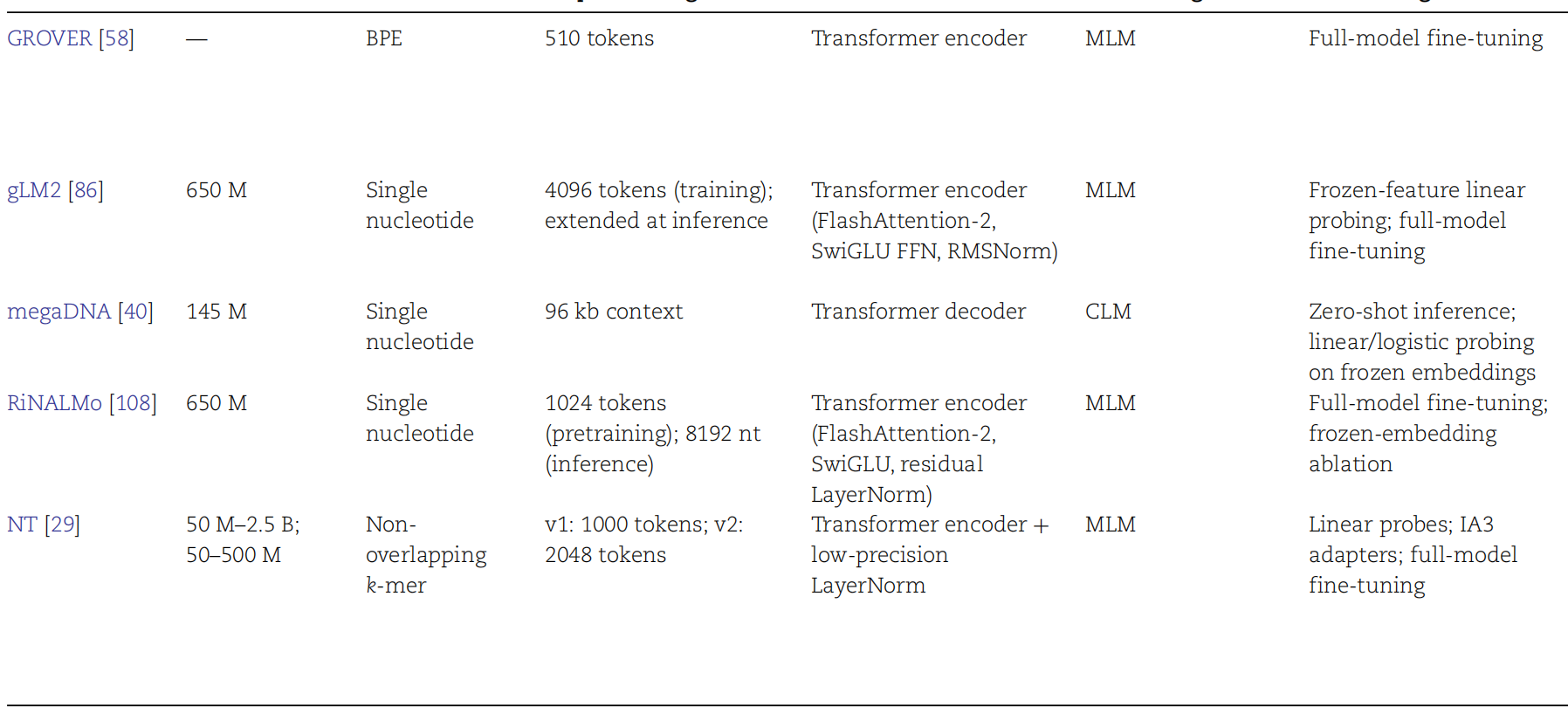 AI Visual Insight: This figure appears to map representative models to tasks, showing which models focus on DNA regulation and which extend to RNA structure or generative design.
AI Visual Insight: This figure appears to map representative models to tasks, showing which models focus on DNA regulation and which extend to RNA structure or generative design.
Developers should prioritize task definition over model popularity in real-world adoption
If the goal is pathogenic variant interpretation, prioritize frameworks that support reference-variant comparison and phenotype regression. If the goal is synthetic design, focus more on conditional generation constraints and experimental feedback loops. In practice, task definition, label quality, sequence length, and evaluation protocol usually matter more than parameter count in determining the upper bound of performance.
FAQ
1. What is the fundamental difference between genome language models and standard NLP Transformers?
Answer: The underlying architecture may look similar, but the input tokens, contextual dependencies, biological priors, and evaluation objectives are fundamentally different. Genomic sequences place much stronger emphasis on long-range dependency, base-level localization, and experimental verifiability.
2. Why is variant effect prediction especially important in this field?
Answer: It directly connects sequence changes to disease risk, expression shifts, and splicing abnormalities, making it one of the most practically valuable tasks in clinical interpretation and functional genomics.
3. How should I choose the right benchmark?
Answer: Start with the task type: classification, regression, or structure prediction. Then check whether it tests cross-species transfer, includes long sequences, and provides high-quality experimental labels. Only after that should you compare model scores.
Core Summary
Based on a review of genome language models, this article reconstructs the core task system for genomic sequence modeling. It covers eight major directions, including regulatory recognition, variant effect prediction, RNA structure, and generative design, while also outlining benchmark priorities and the development path of representative models.